From Inflammation to Scars: Immune-Mediated Fibrogenesis in MASLD
Hello everyone!
Welcome to my blog 🎉
Today, I will describe metabolic dysfunction-associated steatotic liver disease (MASLD) induced fibrosis and what we can learn from it.
Fibrosis: The Point of Maybe Return in MASLD
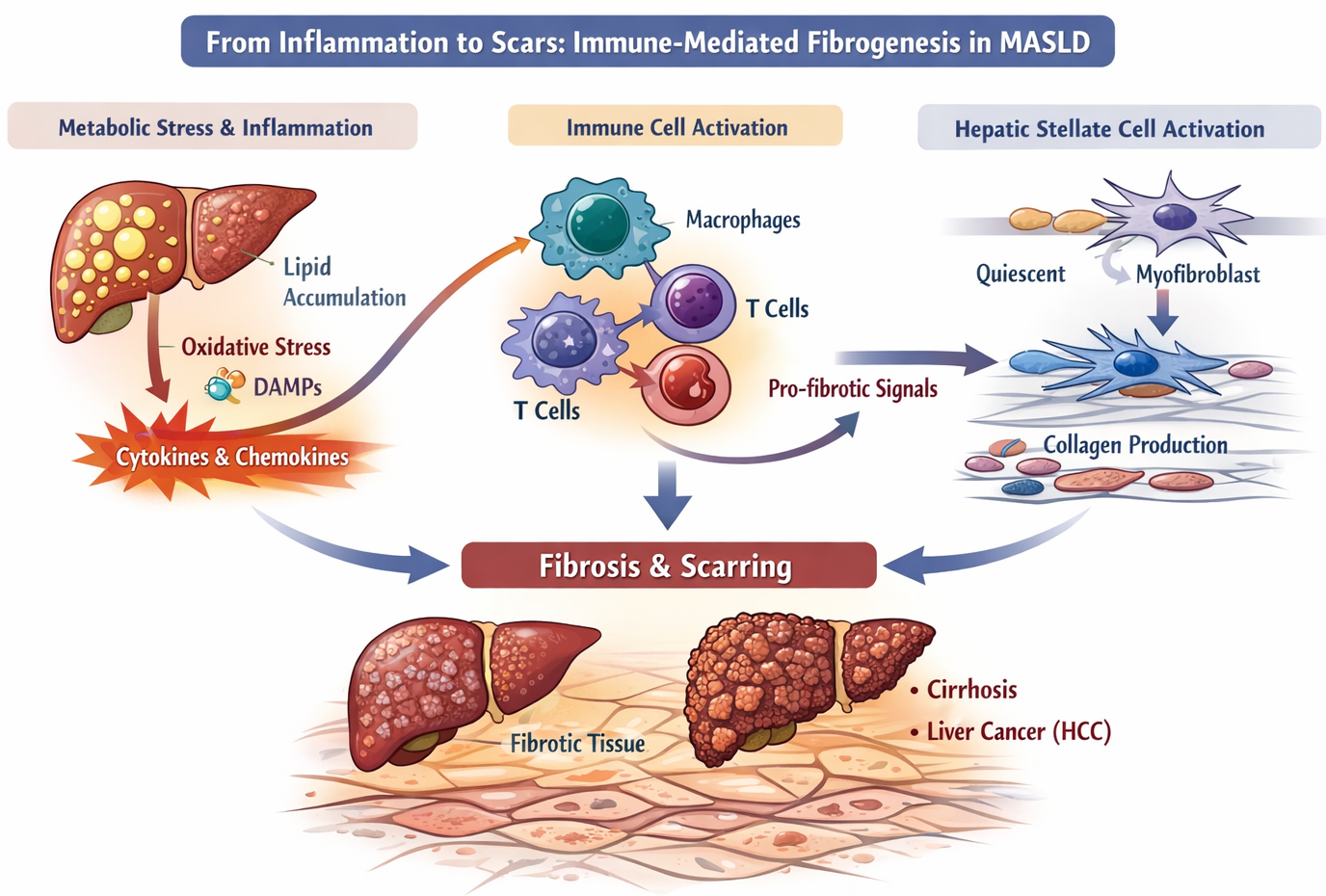
Fibrosis represents the critical pathological turning point in metabolic dysfunction–associated steatotic liver disease (MASLD), transforming a largely reversible metabolic condition into a progressive and potentially life-threatening disorder. While fibrosis is often viewed as a structural or stromal problem, it is, at its core, an immune-mediated outcome of chronic inflammation. The accumulation of scar tissue reflects sustained crosstalk between immune cells and mesenchymal populations, particularly hepatic stellate cells (HSCs), driven by unresolved metabolic stress.
Macrophages as Master Orchestrators of Fibrogenesis
Persistent inflammation creates a cytokine-rich microenvironment that continuously activates immune cells. Among these, macrophages play a central coordinating role. Liver-resident Kupffer cells and recruited monocyte-derived macrophages secrete key profibrotic mediators such as transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), and osteopontin. These signals directly stimulate hepatic stellate cells, initiating their transition from quiescent vitamin A-storing cells into collagen-producing myofibroblasts.
Hepatic Stellate Cells: From Repair to Pathological Scarring
Once activated, hepatic stellate cells become the dominant source of extracellular matrix components, including collagen I and III, fibronectin, and laminin. Crucially, their activation is maintained by ongoing immune input, rather than being a transient wound-healing response. Inflammatory cytokines such as TNF and IL-1β promote stellate cell survival and proliferation, while chemokines produced by activated HSCs recruit additional immune cells, reinforcing a self-perpetuating fibro-inflammatory loop.
Adaptive Immunity Enters the Fibrotic Niche
Beyond innate immune cells, adaptive immune responses increasingly shape fibrosis progression in MASLD. Infiltrating T cells contribute through both direct and indirect mechanisms. T cells interact with hepatic stellate cells, macrophages, and other stromal cells to regulate collagen deposition. These immune-stromal interactions dictate whether fibrosis progresses slowly or rapidly. T cell activation, cytotoxicity, and cytokine production shape the fibrotic response. By studying these cells across liver and adipose tissue, we can uncover mechanisms that might be targeted therapeutically.
I am exploring:
Which T cell subsets correlate with early versus advanced fibrosis?
Can we modulate T cell activity to slow or reverse fibrosis?
Fibrosis as Immunological Memory of Chronic Injury
Fibrosis in MASLD is best understood not as a passive accumulation of scar tissue, but as the immunological imprint of repeated, unresolved metabolic injury. Each fibrotic band reflects cycles of immune activation, hepatocyte damage, and failed repair. The extent of fibrosis correlates strongly with clinical outcomes, including cirrhosis, portal hypertension, and hepatocellular carcinoma, making it the most powerful predictor of disease severity.
Therapeutic Implications: Targeting Immune-Stromal Crosstalk
Viewing fibrosis through an immune-centric lens reframes therapeutic strategy. If fibrosis is driven by persistent immune-stromal interactions, then modulating immune pathways-such as macrophage recruitment, T cell activation, or profibrotic cytokine networks-offers a rational route to altering disease trajectory. In this context, fibrosis is not merely an endpoint of MASLD, but a dynamic and potentially reversible consequence of immune dysfunction.